Recent Highlights
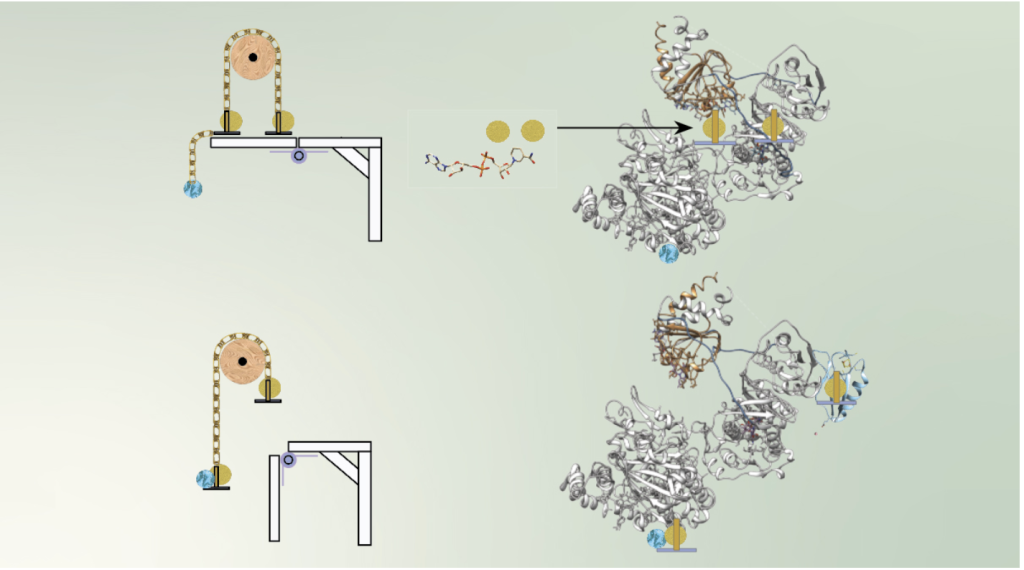
SIBYLS research in Bifurcation of Electrons is highlighted in ALS Science Briefs newsletter
Scientists at SIBYLS used small angle x-ray scattering to understand a microbial protein involved in the bifurcation of high and low energy electrons in microbial metabolism. By analyzing the SAXS[…]
Read more
SIBYLS has opening for Senior Research Associate
This is a wonderful opportunity to work with experienced and innovative scientists in the field of Structural Biology. We are looking for someone with a Bachelor’s Degree (or equivalent knowledge/training)[…]
Read more
Applications of Small Angle Scattering to Structural Biology
SIBYLS beamline scientist, Michal Hammel, partners with BioCAT beamline scientist, Jesse Hopkins to lead a workshop on small angle and neutron scattering at the ACA 74th Annual Meeting in Denver,[…]
Read more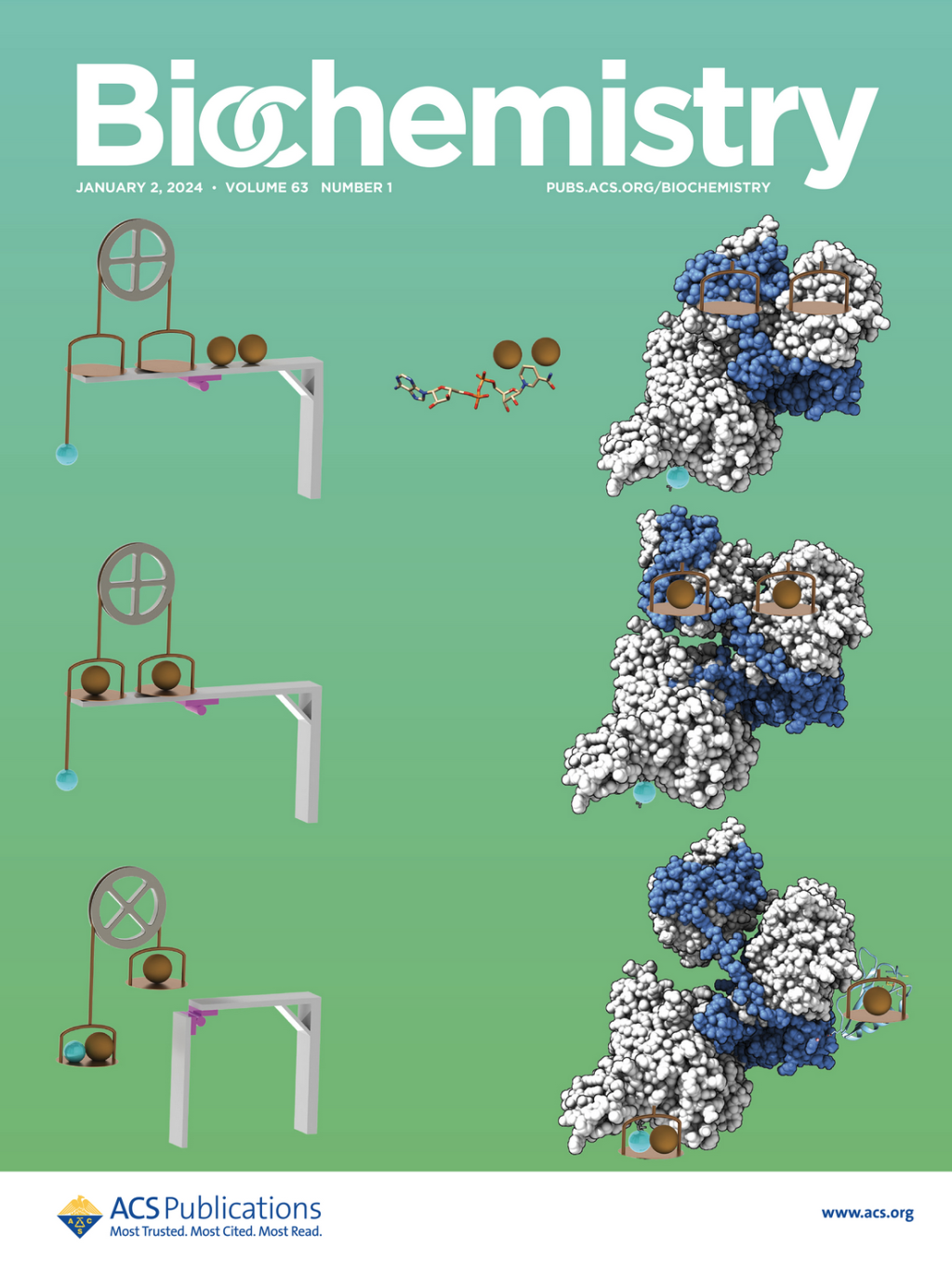
SIBYLS makes cover of Biochemistry
Anaerobic SEC-MALS-SAXS at the SIBYLS beamline of the Advanced Light Source probes the conformational states behind electron bifurcation in the Thermotoga maritima EtfABCX, revealing insights on mechanisms at the thermodynamic[…]
Read more